


阿尔茨海默病是最常见的神经退行性疾病之一。近年来,睡眠—觉醒节律紊乱被认为是阿尔茨海默病早期的重要生物学标志与病理风险因素之一,其出现时间远早于认知障碍等典型临床症状。然而,学界对这一现象涉及的关键脑区与神经机制尚不明晰,制约了阿尔茨海默病早期干预策略的研发进程。
近日,中国科学院上海有机化学研究所生物与化学交叉研究中心研究团队,系统揭示了阿尔茨海默病早期睡眠障碍的潜在机制。
研究团队通过5xFAD转基因阿尔茨海默病小鼠模型,结合睡眠监测、全细胞膜片钳、药理学调控等多种技术手段发现,2月龄阶段的5xFAD小鼠已出现暗周期特异性觉醒过度及脑状态转换能力下降的病理表型。在机制层面,研究团队观察到蓝斑神经元存在暗周期时相特异性自发放电频率升高的现象,其底层机制表现为:Aβ寡聚体干扰了维持蓝斑神经元正常活动水平的α2A型去甲肾上腺素受体功能,使其对两种具有神经元活性“刹车”效应的电压门控钾离子通道4(Kv4)与电压门控钾离子通道7(Kv7)的调节功能失灵,进而造成蓝斑神经元兴奋性在暗周期出现异常升高。
研究进一步显示,采用药理学手段激活α2A型去甲肾上腺素受体或增强Kv7通道功能,可有效恢复蓝斑神经元的正常电活动,并明显改善阿尔茨海默病小鼠的睡眠—觉醒节律紊乱,使其恢复至正常水平。
该研究揭示了阿尔茨海默病早期睡眠障碍的关键异常脑区与潜在机制,并提示α2A型去甲肾上腺素受体与Kv7通道可作为早期干预的潜在治疗靶点,为研发针对阿尔茨海默病早期睡眠症状的精准治疗策略提供了新思路。
相关研究成果发表在Alzheimer's & Dementia上。研究工作得到国家自然科学基金委员会、上海市等的支持。
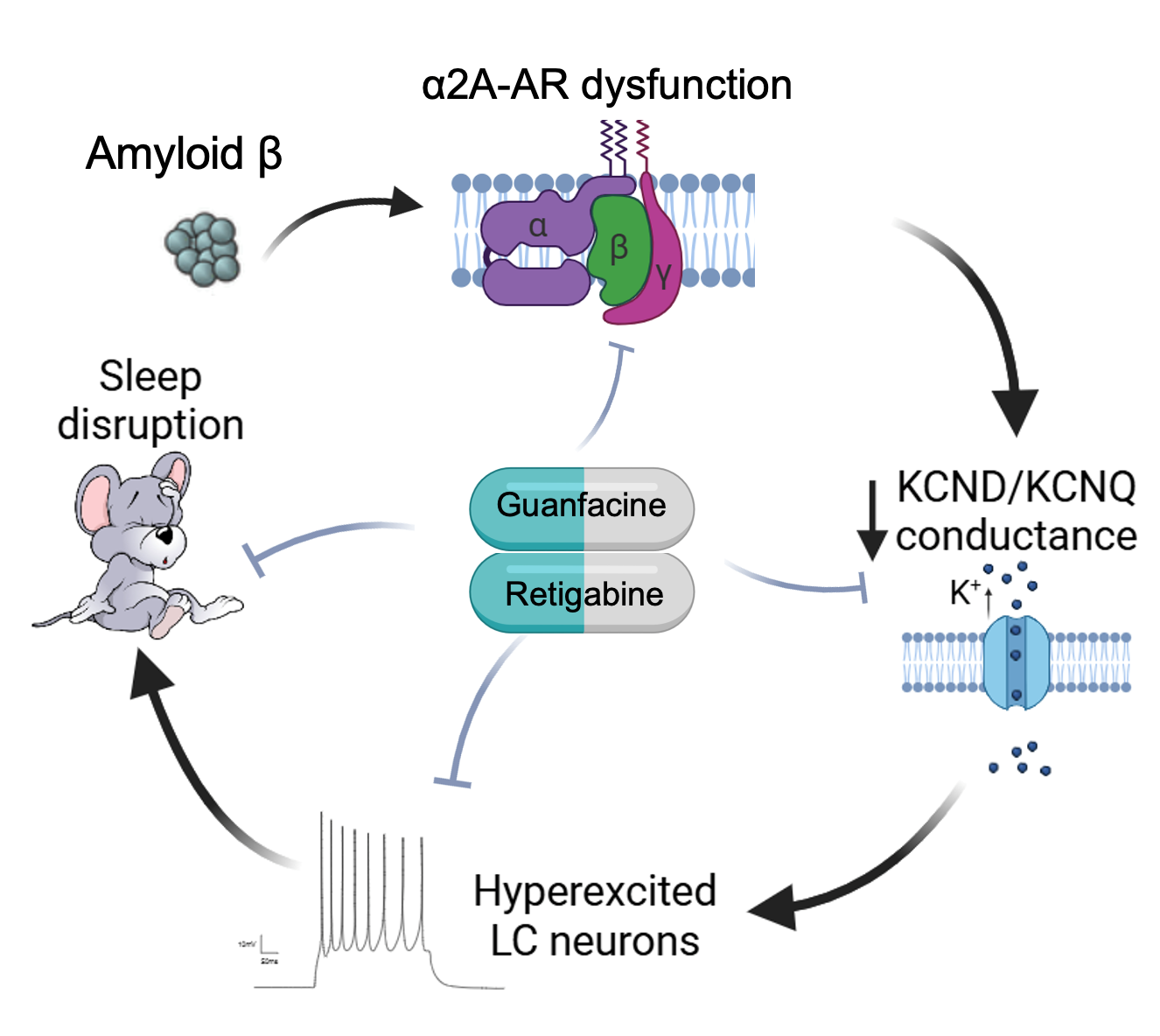
阿尔茨海默病早期睡眠障碍机制的工作模型









